What is Pharmacovigilance?
The word Pharmacovigilance can be divided into ‘Pharmakon’ and ‘Vigilance’. In Greek
Pharmakon means ‘a drug or medicine’ and the word ‘Vigilance’ has been derived from
a latin word ‘Vigilare’, which means ‘to watch’.
WHO has defined pharmacovigilance as “the science and activities relating
to the detection, assessment, understanding and prevention of adverse effects or
any other drug-related problem” – with drug-related problems being further defined
as “issues that affect the safety and safe use of medicines.”
Goal of pharmacovigilance is to foster the rational and safe use of medicines.
Pharmaco = Medicine
Vigilare = To watch
WHO Definition of PV:
The science and activities relating to the detection, assessment, understanding
and prevention of adverse effects or any other drug-related problem.
Two stages
- PRE MARKETING
- POST MARKETING
"Dying from a disease is sometimes unavoidable. But, dying from an adverse drug reaction
is unacceptable".
-Dr Vladimir Lepakhin
Geneva 2005
The information collected during the pre-marketing phase of a medical drug is inevitably
incomplete with regard to possible adverse reactions:
Tests in animals are insufficiently predictive of human safety.
Example:Practolol, a ß1-adrenoreceptor blocking agent withdrawn from the U.K.
market in 1976 after several years of widespread use. The U.K. action was prompted
by the serious adverse reactions of dermatitis, keratoconjunctivitis and sclerosing
peritonitis, collectively termed the oculomucocutaneous syndrome. This syndrome
had not been seen during extensive preclinical animal testing conducted within required
guidelines.
In clinical trials patients are selected and limited in number, the conditions of
use differ from those in clinical practice and the duration of trials is limited.
At least 30,000 people need to be treated with a drug to be sure that you do not
miss at least one patient with an ADR which has an incidence of 1 in 10,000 exposed
individuals. In order to have a 95% chance of detecting an adverse event with an
incidence of 1 per 1,000, 3,000 patients at risk are required; with no more than
3,000 to 4,000 individuals usually exposed to a medical product prior to marketing.
Information about rare but serious adverse reactions, chronic toxicity, use in special
groups (such as children, the elderly or pregnant women) or drug interactions is
often incomplete or not available.
Ethical concern: Not reporting a serious unknown reaction is unethical. ‘To know
of something that is harmful to another person, who does not know, and not telling,
is unethical’.
Cost: In USA over 770,000 people are injured or die each year in hospitals from
adverse drug events (ADEs), which may cost up to $5.6 million each year per hospital
depending on hospital size. the country of origin of the drug. Pharmacovigilance
is needed for the prevention of drug-induced human suffering and to avoid financial
risks associated with unexpected adverse effects.
SOME IMPORTANT DATES IN THE HISTORY OF PHARMACOVIGILANCE
1848, The Lancet starts collecting notifications of side effects after a death caused
by anaesthesia.
1906, US Federal Food and Drug Act requires that pharmaceuticals be “pure” and “free
of any contamination”.
1937, USA: 107 lethal cases after diethylenglycol was mistakenly used to solubilize
sulphanilamides.
1952, France: 100 lethal cases after diethyl tin diodide was mistakenly used in
a skin preparation
1961, Dr William McBride (Australia) reported 20% increase in foetal abnormalities
and significant increase of phocomelia in relation with thalidomide use, later numerous
reports from other countries (more than 4000 cases).
1962, USA Kefauver-Harrise amendment to the law (requirement to prove safety and
efficacy before issuing Marketing Authorization).
1964, UK starts “yellow cards” system.
1967, Start of WHO Programme for International Drug Monitoring.
1976, Drugging of the Americas: inadequacy of safety information.
1990 , ICH - elaboration of intra-regional requirements for safety starts
The events such as sulphanilamide, chloramphenicol and thalidomide tragedies are
best regarded as precipitating jolts, that define the significance of safety updates
and trigger an evolution of regulatory logics.
Why Pharmacovigilance is needed in every country?
Differences among countries (and even regions within countries) in the occurrence
of ADRs and other drug-related problems.
Examples:
Diseases and prescribing practices;
Genetics, diet, traditions of the people;
Drug manufacturing processes used which influence pharmaceutical quality and composition;
Drug distribution and use including indications, dose and availability;
The use of traditional and complementary drugs (e.g. herbal remedies) which may
pose specific toxicological problems, when used alone or in combination with other
drugs.
Aims of Pharmacovigilance:
To enhance patient care and patient safety in relation to the use of medicines,
especially with regard to the prevention of unintended harm from the use of drugs
To improve public health and safety in relation to the use of medicines by the provision
of reliable, balanced information resulting in more rational use of drugs;
To contribute to the assessment of the risk-benefit profile of medicines,
To encourage safer and more effective use of medicines
Promote understanding, education and clinical training in Pharmacovigilance and
it’s effective communication to the public.
Key Terminologies
Adverse Event (AE)
An adverse event is any untoward medical occurrence in a patient administered a
medicinal product and which does not necessarily have to have a causal relationship
with this treatment. An adverse event can therefore be any unfavorable and unintended
sign (for example, an abnormal laboratory finding), symptom, or disease temporally
associated with the use of a medicinal product, whether or not considered related
to this medicinal product.
Synonym: adverse experience.
Adverse Drug Reaction (ADR)
A response which is noxious and unintended, and which occurs at doses normally used
in humans for the prophylaxis, diagnosis, or therapy of disease, or for the modification
of physiological function (WHO 1972).
Minimum Information to Report an SAE
Identifiable reporting source (reporter)
Identifiable patient
Name of a suspect medicinal product
Event that can be identified as serious (missing details can be reported later)
Healthcare Professional:
Healthcare professional is defined as a medically-qualified person such as a physician,
dentist, pharmacist, nurse, coroner, or as otherwise specified by local regulations.
Consumer:
Consumer is defined as a person who is not a healthcare professional such as a patient,
lawyer, friend, or relative of a patient.
Clinical Trial:
A systematic study on pharmaceutical products in human subjects (including patients
and other volunteers) in order to discover or verify the effects of and/or identify
any adverse reaction to investigational products, and/or to study the absorption,
distribution, metabolism and excretion of the products with the objective of ascertaining
their efficacy and safety .
Dechallenge
The withdrawal of a drug from a patient; the point at which the continuity, reduction
or disappearance of adverse effects may be observed.
Rechallenge
The point at which a drug is again given to a patient after its previous withdrawal.
Post-marketing
The stage when drug is generally available on the market.
Spontaneous reports
A spontaneous report (SR) is an unsolicited communication by healthcare professionals
or consumers to a company, regulatory authority or other organization (e.g. regional
centers, poison control center) that describe one or more adverse drug reactions
in a patient who was given one or more medicinal products and that does not derive
from a study or any organized data collection scheme.
Periodic safety Update Report (PSUR)
PSURs present worldwide safety experience of a medicinal product at defined times
post-authorization. PSUR includes all relevant new safety information from appropriate
sources and relate these data to patient exposure. It also indicates whether changes
will be made to product information in order to optimize the use of the product.
Benefit and Risk Analysis
Examination of the favorable (beneficial) and unfavorable results of undertaking
a specific course of action.
Pharmacovigilance Process
Good Pharmacovigilance Practices
Spontaneous reports have been the historical cornerstone of the pharmacovigilance
component of PMS. The scientifically supported maximal use of these reports is a
central goal of the pharmacovigilance. Good pharmacovigilance processes (GPVP) focus
on the enhancement of the reports that are most likely to be important. In parallel
to any regulatory reporting (submission) triage these reports need to have both
their potential maximized and the value measured. That measurement must be both
as an individual and in a case-series. All activity must be well documented.
The goal of GPVP is to clearly and accurately identify rare, serious, unusual or
unexpected adverse drug reactions as soon as possible after market launch. This
plays to the few strengths inherent in this type of anecdotal observational data.
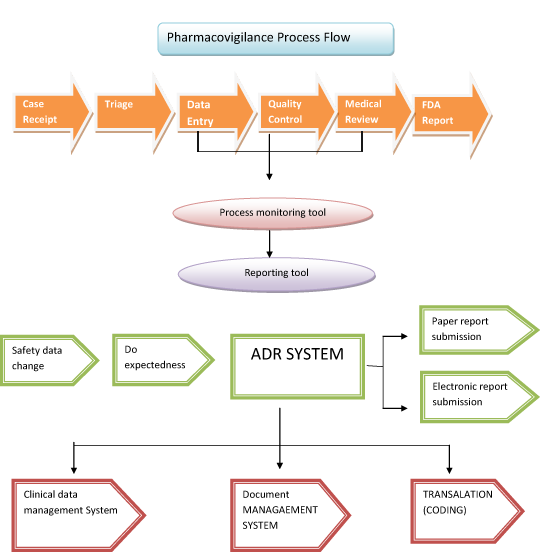
Spontaneous reports are proposed as a step-wise approach:
- step 1 – Case receipt
- step 2 – Triage
- step 3 – Data Entry
- step 4 – Quality Control
- step 5 – Medical Review
- step 6 – FDA Report Submission
Pharmacovigilance Laws & Regulations
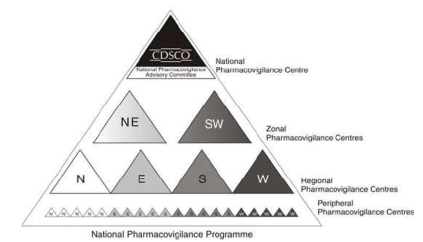
Pharmacovigilance in India
History: National Pharmacovigilance Advisory Committee (NPAC) was created
under the chairmanship of the Director General of Health Services and Drug Controller
General of India (DCGI).
National Pharmacovigilance Programme for India is sponsored by the World Health
Organization (WHO) and is funded by the World Bank.
European Union:
Directive 2001/83, amended by Directive 2004/27. This concerns all medicinal products,
although for Pharmacovigilance it is most relevant to products authorized by
- The national procedure
- Mutual recognition
- Centralized procedures.
The Member States are the licensing authorities in these procedures.
The principle guidance documents are summarized in Volume 9A of “The rules governing
medicinal products in the European Union – Pharmacovigilance” (Volume 9A)and in
the pharmacovigilance related guidelines of ICH (E2 series) which incorporates international
agreements reached within the framework of the International Conference on Harmonisation
(ICH).The European commission (EC) is responsible for setting legislation and corresponding
guidance for those countries in Europeanunion (EU).
The EU phvg system is called as Eudravigilance.Therefore EC is responsible for EMEA.
National Authorities – each country’s CA is responsible for monitoring the
safety profile of products in its territory and taking action when necessary.
European Commission - is responsible for overall community system of PV and
legal framework(authorization).
EMEA - evaluates and supervises medicinal products in the EU and provision
of advise on measures to ensure safety and effective use.
Japan - The regulatory authority in Japan is Ministry of health, labor and
welfare (MHLW).Recently there is an increase ADR’s reported in Japan.
Spontaneous reporting system (SRS) in Japan - Spontaneous reporting system
SRS was created in 1967 in the early stage of Japan. The reports were sent from
the designated medical institute to MHLW.Good vigilance practiced (GVP) and the
adverse drug reaction reports are reported to the companies (MAH).The good vigilance
practiced and the good quality practiced (GQP) represents the license rules that
the MAH must follow.
USA - The regulatory authority in US is Food and Drug Administration and
the guidance document is Code of federal regulations. i.e. CFR part 21 was established
in 1930.It is an agency of the United States department of health and human services.AERS
for pharmacovigilance in US.
Medwatch program for reporting the ADR’s.(mandatory and voluntary) and NDA annual
reports are submitted to FDA.Consumer reports and the dear HCP letters.A fifteen
day report submitted during the reporting period.A Med watch form for each adverse
event not previously submitted as a fifteen day report.form 3500A – mandatory and
for MAH use. form 3500- voluntary and for HCP and consumer use.